

mRNA injections are genetically modified organisms - the impact on future generations is unknown - you may need to expand your email!

With the authors kind permission here is a verbatim reproduction of a rigorously reviewed and painstakingly researched paper on how Pfizer and Moderna were allowed to bypass EU and Australian regulations.
Each sentence and paragraph carries too much significance to be abridged or summarized without losing sight of the whole.
Here are a couple of excerpts:
“..clearly satisfy deeming the LNP-modRNAs contained in the Pfizer and Moderna products as Genetically Modified Organisms.”
GMO’s.
The paper reprises research on “Retroposition” and has this comment:
“.. The mechanism that leads to the formation of retrocopies in a human lineage is relatively well studied and predominantly includes Long Interspersed Element-1 (L1/LINE-1) retrotransposons”
The investigatory paper also touches on the hereditary impacts of the use of GMO’s in the injections and the denial of informed consent to clinical trial participants.
Here is the paper.
The Canaries in the Human DNA Mine
An investigation
by Julian Gillespie LLB, BJuris
8 March 2023
On or about 17 July 2020 the following regulation came into force across the European Union:
REGULATION (EU) 2020/1043 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL
Yes, the important words to note are:
genetically modified organisms (GMOs)
For simplicity let us call the above Regulation 2020/1043. For further simplicity, Regulation 2020/1043 did not want you to know about any GMOs contained in Covid-19 drugs.
Side note: I wonder if any attempts were made by Australian regulators to conceal GMOs contained in Covid-19 drugs? I will address that specific topic after we walk through what the Europeans did first.
Before diving into the reeds of Regulation 2020/1043, an understanding of two prior and extremely important European Parliament Directives impacted by Regulation 2020/1043 is required, being Directive 2001/18/EC and Directive 2009/41/EC - two Directives meant to protect human life.
Note: what follows is a deep dive into European Union legislation, which from any reasonable view appears to have been made purposefully difficult to navigate, due to constant references to other Directives and Regulations, which all require reading before the plain intent and reach of any immediate statute under review can be arrived at. This paper has gone down the rabbit hole of European GMO laws with the aim being to rip away what appears to be laws used as wallpaper, in an attempt to conceal the truth from European citizens, and in turn, the world.
Directive 2001/18/EC
Notification Information Required
before the Deliberate Release of GMOs into the Environment
or when seeking Authorisation for GMO products to Enter the Market
Sponsors in the EU who wish to release or introduce a product containing GMOs must notify relevant EU regulatory authorities pursuant to Directive 2001/18/EC, where the Objective of the Directive is unambivalent:
In accordance with the precautionary principle, the objective of this Directive is to approximate the laws, regulations and administrative provisions of the Member States and to protect human health and the environment when:
- carrying out the deliberate release into the environment of genetically modified organisms for any other purposes than placing on the market within the Community,
- placing on the market genetically modified organisms as or in products within the Community.
The definition for what constitutes a GMO is found at Article 2 (emphasis added):
For the purposes of this Directive:
(1) "organism" means any biological entity capable of replication or of transferring genetic material;
(2) "genetically modified organism (GMO)" means an organism, with the exception of human beings, in which the genetic material has been altered in a way that does not occur naturally by mating and/or natural recombination;
Within the terms of this definition:
(a) genetic modification occurs at least through the use of the techniques listed in Annex I A, Part 1
Annex I A provides a non-exhaustive list of techniques for genetic modification. For now, note the technique described at Annex I A, Part 1(2):
TECHNIQUES REFERRED TO IN ARTICLE 2(2)
PART 1
Techniques of genetic modification referred to in Article 2(2)(a) are inter alia:
(2) techniques involving the direct introduction into an organism of heritable material prepared outside the organism including micro-injection, macro-injection and micro-encapsulation;
Some further definitions under Article 2 are noteworthy for gaining context:
(3) "deliberate release" means any intentional introduction into the environment of a GMO or a combination of GMOs for which no specific containment measures are used to limit their contact with and to provide a high level of safety for the general population and the environment;
(4) "placing on the market" means making available to third parties, whether in return for payment or free of charge;
(5) "notification" means the submission of the information required under this Directive to the competent authority of a Member State;
(6) "notifier" means the person submitting the notification;
(7) "product" means a preparation consisting of, or containing, a GMO or a combination of GMOs, which is placed on the market;
(8) "environmental risk assessment" means the evaluation of risks to human health and the environment, whether direct or indirect, immediate or delayed, which the deliberate release or the placing on the market of GMOs may pose and carried out in accordance with Annex II.
And to be properly technical, ‘environment’ does not mean just the great outdoors .. forests, lakes, creeks, and streams .. the term also includes the human body.
Moving to Article 4 and we start to appreciate Directive 2001/18/EC is for safeguarding human health from GMOs. Article 4 reads in part (emphasis added):
1. Member States shall, in accordance with the precautionary principle, ensure that all appropriate measures are taken to avoid adverse effects on human health .. which might arise from the deliberate release or the placing on the market of GMOs. GMOs may only be deliberately released or placed on the market in conformity with part B or part C respectively.
2. Any person shall, before submitting a notification under part B or part C, carry out an environmental risk assessment. The information which may be necessary to carry out the environmental risk assessment is laid down in Annex III. Member States and the Commission shall ensure that GMOs which contain genes expressing resistance to antibiotics in use for medical or veterinary treatment are taken into particular consideration when carrying out an environmental risk assessment, with a view to identifying and phasing out antibiotic resistance markers in GMOs which may have adverse effects on human health and the environment. This phasing out shall take place by the 31 December 2004 in the case of GMOs placed on the market according to part C and by 31 December 2008 in the case of GMOs authorised under part B.
The bolded section in Article 4(2) takes on particular relevance with respect to the modRNA sequencing mechanism in the Pfizer and Moderna Covid-19 drugs. In the article Curious Kitten, aka, Deep sequencing of the Moderna and Pfizer bivalent vaccines identifies contamination of expression vectors designed for plasmid amplification in bacteria, a team led by Kevin McKernan demonstrates that the 2022/23 bivalent Covid-19 drugs are contaminated with double stranded Plasmid DNA (dsDNA), with the Pfizer product heavily contaminated with ‘billions of antibiotic resistant plasmids injected per person per shot’.
But .. the manufacturing techniques used for the bivalent ‘vaccines’ is said to be a mirror of the manufacturing techniques Pfizer and Moderna created in 2020 for their monovalent ‘vaccines’. Manufacturing techniques are meant to get better with time and practice. Should we wonder what the contaminate levels were/are like in the monovalent ‘vaccines’ that were shot into the arms of billions of people? And if a private citizen (scientist) could confirm literally tons of contaminates, then if European regulators missed that, what other stuff did they miss or know about in the monovalent ‘vaccines’, but have not informed folks about?
But I digress.
Part B of Directive 2001/18/EC contains Article 5 through Article 11, and concerns the GMO information that a sponsor must submit as part of an Environmental Risk Assessment, for “ANY OTHER PURPOSE THAN FOR PLACING ON THE MARKET”.
In short, Part B applies to any sponsor seeking the deliberate release of GMOs into the environment, be it an airborne release or release into waters or a release into soils. As ‘environment’ includes the human body, a ‘deliberate release’ can include the intentional release of GMOs across an entire population, where the intended ‘receiving environments’ for a GMO are the bodies of those persons receiving the GMO, by way of injection for example.
Article 5 does state:
Articles 6 to 11 shall not apply to medicinal substances and compounds for human use
However that caveat only applies when other legislation authorised by the European Community is in place and requires sponsors to provide, prior to the grant of any consent, all the GMO information detailed in Article 5(1)(a), (b), (c), and (d). No such other legislation exists, therefore Articles 6 through 11 also apply to ‘medicinal substances and compounds for human use’.
Article 6 stipulates sponsors must supply a comprehensive dossier of information addressing all the items set forth in Annexures II and III. I will address those information requirements further below, as they also apply to Part C of the same Directive.
Part C of Directive 2001/18/EC contains Articles 12 to 24, which detail the even more stringent GMO information a sponsor must provide as part of an Environmental Risk Assessment, when “PLACING ON THE MARKET OF GMOs AS OR IN PRODUCTS”.
Articles 12, 13, and 14 stipulate sponsors must supply a comprehensive dossier of information addressing all the items set forth in Annexures II, III, IV, VII. Information required under Annex VII is only required after a competent authority [a medicines/drug regulator] has reviewed the information received pursuant to Annexures II, III, and IV, and that authority has returned a favourable Assessment Report (Article 14(3)(a)) to the sponsor, containing the information and conclusions as set forth under Annex VI.
Directive 2001/18/EC is important to folks everywhere when we reach Articles 13 through 24. The Part B Articles 5 through 11 are of equal import, but for reasons that will become apparent later, focusing on Articles 13 through 24 is the better course for what is clearly a long note being shared here.
Article 13 states (in part, emphasis added):
Notification procedure
1. Before a GMO or a combination of GMOs as or in products is placed on the market, a notification shall be submitted to the competent authority of the Member State where such a GMO is to be placed on the market for the first time.
The competent authority shall without delay examine whether the notification is in accordance with paragraph 2 and shall, if necessary, ask the notifier for additional information.
2. The notification shall contain:
(a) the information required in Annexes III and IV. This information shall take into account the diversity of sites of use of the GMO as or in a product and shall include information on data and results obtained from research and developmental releases concerning the impact of the release on human health and the environment;
(b) the environmental risk assessment and the conclusions required in Annex II, section D;
(e) a plan for monitoring in accordance with Annex VII, including a proposal for the time-period of the monitoring plan; this time-period may be different from the proposed period for the consent;
(f) a proposal for labelling which shall comply with the requirements laid down in Annex IV. The labelling shall clearly state that a GMO is present. The words "this product contains genetically modified organisms" shall appear either on a label or in an accompanying document;
Returning to the term ‘environment’ also being inclusive of the human body, when a GMO is contained in a product designed to be released into the environments of many human bodies, Article 13(2) requires the sponsor of that product take into account the diversity of human bodies. That is a pretty big ask when it comes to national populations across Europe. As for the notification information having to include the impact of the GMO on human health, are we not talking about genetically modified organisms here, capable of transferring genetic material, where that Genetically Modified Organism is composed of ‘heritable material’? The question becomes then: how long would be needed for researchers to obtain adequate data in respect of the ‘heritable material’ effects of a GMO? A few months? A couple of years? But inheritance of genetic material requires generational studies looking at how any children born of those who received a GMO into their body, fare in respect of their development as offspring who inherited the new ‘heritable material’. For mine, I could not be assured by any data generated by a sponsor after only a few short months concerning offspring still not born, Are you?
Before moving on I will just ask in respect of Article 13(2)(f) whether anyone has seen any Covid-19 vaccine products labelled with “this product contains genetically modified organisms”? No one? .. me neither.
Article 13 requires a sponsor of a GMO product to provide a ton of information. A ton. Annexures II, III, IV, and VII take some time to scroll through when viewing Directive 2001/18/EC, with each annexure seeking to ensure that when it comes to GMOs, every possible harm to human health are examined and presented in detail by the sponsor to regulators, and in turn the public, prior to any approval of their products.
Annex II really sets the stage for how impactful Genetically Modified Organisms can be. When reading some of Annex II reproduced below, think of any drug containing GMOs and think of how far into the future their effects could be:
ANNEX II
PRINCIPLES FOR THE ENVIRONMENTAL RISK ASSESSMENT
This Annex describes in general terms the objective to be achieved, the elements to be considered and the general principles and methodology to be followed to perform the environmental risk assessment (e.r.a.) referred to in Articles 4 and 13.
With a view to contributing to a common understanding of the terms "direct, indirect, immediate and delayed" when implementing this Annex, without prejudice to further guidance in this respect and in particular as regards the extent to which indirect effects can and should be taken into account, these terms are described as follows:
- "direct effects" refers to primary effects on human health or the environment which are a result of the GMO itself and which do not occur through a causal chain of events;
- "indirect effects" refers to effects on human health or the environment occurring through a causal chain of events, through mechanisms such as interactions with other organisms, transfer of genetic material, or changes in use or management.
Observations of indirect effects are likely to be delayed;
- "immediate effects" refers to effects on human health or the environment which are observed during the period of the release of the GMO. Immediate effects may be direct or indirect;
- "delayed effects" refers to effects on human health or the environment which may not be observed during the period of the release of the GMO, but become apparent as a direct or indirect effect either at a later stage or after termination of the release.
A general principle for environmental risk assessment is also that an analysis of the "cumulative long-term effects" relevant to the release and the placing on the market is to be carried out. "Cumulative long-term effects" refers to the accumulated effects of consents on human health and the environment,
Annex II goes on to stipulate many other issues and topics that MUST be addressed in an Environmental Risk Assessment (ERA), required to be prepared by a sponsor, amounting to a huge bundle of materials that MUST be submitted by a sponsor for consideration by EU State Members, BEFORE marketing authorisation can be considered for the product containing the GMOs.
A small set of the Environmental Risk Assessment information items required include (with some paraphrasing, emphasis added, comments in [] mine):
· Any characteristics of the GMOs linked to the genetic modification that may result in adverse effects on human health
· Identifying the particular potential adverse effects arising from the genetic modification
· Potential adverse effects of from the GMOs including:
- disease to humans including allergenic or toxic effects;
- effects on the dynamics of populations of species in the receiving environment [humans];
- altered susceptibility to pathogens facilitating the dissemination of infectious diseases;
- compromising prophylactic or therapeutic medical .. protection treatments, for example by transfer of genes conferring resistance to antibiotics used in human medicine.
· Adverse effects may occur directly or indirectly through mechanisms which may include:
- the spread of the GMO(s) in the environment [within the human body and outside];
- the transfer of the inserted genetic material to other organisms [to the recipient and/or their offspring];
- phenotypic and genetic instability.
Annex II(C.2)(2) goes further and implicitly invokes a precautionary principle approach – and recall, ‘environment’ includes the human body:
· The magnitude of the consequences of each potential adverse effect should be evaluated.
· This evaluation should assume that such an adverse effect will occur. The magnitude of the consequences is likely to be influenced by the environment into which the GMO(s) is (are) intended to be released and the manner of the release.
For clarity, Annex II(D) further stipulates a range of issues needing to be addressed, including:
· Potential immediate and/or delayed environmental impact of the direct and indirect interactions between the GMO and target organisms.
.. where the ‘target organism’ in the case of a medicine containing GMOs would of course be, receiving humans.
Complimenting Annex II are the further information requirements detailed under Annex III, some of which include:
· classification of hazard according to existing Community rules concerning the protection of human health
· information on survival
· stability of the organism in terms of genetic traits
· rate and level of expression of the new genetic material
· toxic or allergenic effects of the GMOs
· if the organism is pathogenic to humans who are immunocompetent:
- diseases caused and mechanism of pathogenicity including invasiveness
- antibiotic resistance patterns
- allergenicity
· genetic transfer capability:
- post release transfer of genetic material from GMOs into organisms [humans]
· measures employed to ensure and to verify genetic stability. Description of genetic traits which may prevent or minimise dispersal of genetic material. Methods to verify genetic stability
· methods for tracing the GMOs, and for monitoring their effects
· techniques for detecting transfer of the donated genetic material to other organisms [babies?]
· plans for protecting human health .. in case of the occurrence of an undesirable effect.
Annex IV concerns additional information requirements needed by regulators in a post-marketing approval environment, for storage and handling and packaging and labelling, not least of which being labels clearly marked with: "This product contains genetically modified organisms".
Pursuant to Article 13 and the equally important issue of Monitoring effects from the GMOs in a post-marketing approval environment are detailed under Annex VII, where the stated Objective of Monitoring is sensible and to the point:
· confirm that any assumption regarding the occurrence and impact of potential adverse effects of the GMO or its use in the Environmental Risk Assessment are correct, and
· identify the occurrence of adverse effects of the GMO or its use on human health or the environment which were not anticipated in the Environmental Risk Assessment.
Further, (this is reproduced for tragic irony), and the design of the monitoring plan should:
· give consideration to the mechanisms for identifying and confirming any observed adverse effects on human health .. and enable the consent holder [sponsor/manufacturer] or the competent authority [regulatory authority who approved the GMOs], where appropriate, to take the measures necessary to protect human health ...
To this point it is abundantly clear that the information required from sponsors of GMO products is, extensive. After all the Objective of Directive 2001/18/EC requires the strict observance of the precautionary principle for the purpose of protecting human health.
Recall that Part B of Directive 2001/18/EC covering Articles 5 through 11 applies to any sponsor seeking the deliberate release of GMOs into the ‘environment’, where environment includes individual human bodies. Though Part B is not as burdensome as Part C when a sponsor seeks approval for their GMO product to go into the market, Part B nonetheless commands the provision of extremely detailed information under Annexures II and III, being an Environmental Risk Assessment and information specifically about the GMO in question. So Part B is also concerned with the same Objective requiring the strict observance of the precautionary principle for the purpose of protecting the health of humans who may receive the GMO into their environment, their body.
Lastly, and perhaps most importantly, the GMO information requirements needing to be submitted by way of a Notification to appropriate European authorities, must also be presented to the general public.
Part B states in clear terms under Article 9 (for GMO release into the environment):
Consultation of and information to the public
1. Member States shall .. consult the public and, where appropriate, groups on the proposed deliberate release. In doing so, Member States shall lay down arrangements for this consultation, including a reasonable time-period, in order to give the public or groups the opportunity to express an opinion.
Part C states in clear terms under Article 24 (for GMO products entering the market):
Information to the public
.. upon receipt of a notification in accordance with Article 13(1), the Commission shall immediately make available to the public the summary referred to in Article 13(2)(h). The Commission shall also make available to the public assessment reports in the case referred to in Article 14(3)(a). The public may make comments to the Commission within 30 days. The Commission shall immediately forward the comments to the competent authorities.
.. for all GMOs which have received written consent for placing on the market .. the assessment reports carried out for these GMOs and the opinion(s) of the Scientific Committees consulted shall be made available to the public. For each product, the GMO or GMOs contained therein and the use or uses shall be clearly specified.
Directive 2009/41/EC
Notification Information Required before Contained Use of GMOs
As mentioned in the opening, Regulation 2020/1043 of 17 July 2020 also impacted the operation of Directive 2009/41/EC.
Directive 2009/41/EC is all about Contained Use. Specifically, the Directive speaks to the use of GMOs in a laboratory environment, the risks associated when experimenting with GMOs, and the need to properly quantify those risks and place appropriate controls around them, chiefly through the use of differing levels of containment in response to the risks identified – Class1, Class 2, Class 3, or Class 4 – where Class 1 activities involve no or negligible risk, up to Class 4 involving high risk where a Level 4 Containment facility is required to protect human health and the environment.
The definitions for what are deemed to be a ‘micro-organism’ and a ‘genetically modified micro-organism’ (GMM) mirror the definition for a ‘genetically modified organism’ seen with Directive 2001/18/EC above.
Articles 4 through 13 involve the Notification information that must be submitted before the contained use of a GMO can commence. A great deal of reference is made to the numerous Annexures which detail the manner for undertaking a risk assessment of the GMOs, and the appropriate risk Class and Containment Level to be assigned to those identified risks, and the regulatory response required from the designated ‘competent authority’ responsible for granting permission for the contained use to commence.
The absolute importance of the risk assessment required, the information required prior to approval, and the involvement of regulators in that approval process cannot be underscored enough. An example of the threat to human life when dealing with GMOs in a contained environment is brought home to the reader, when one looks at some of the containment measures required under Annex IV:
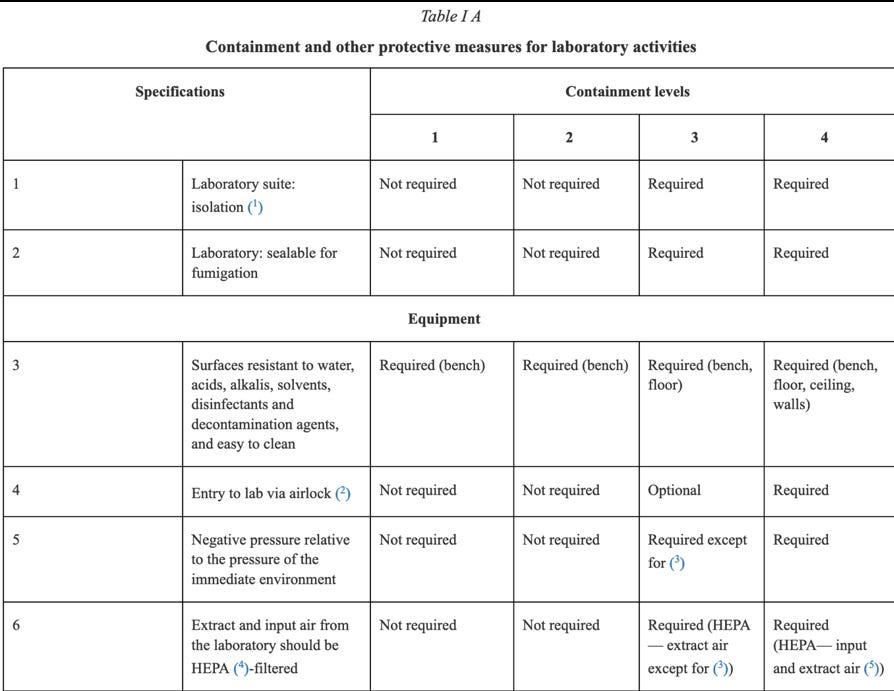
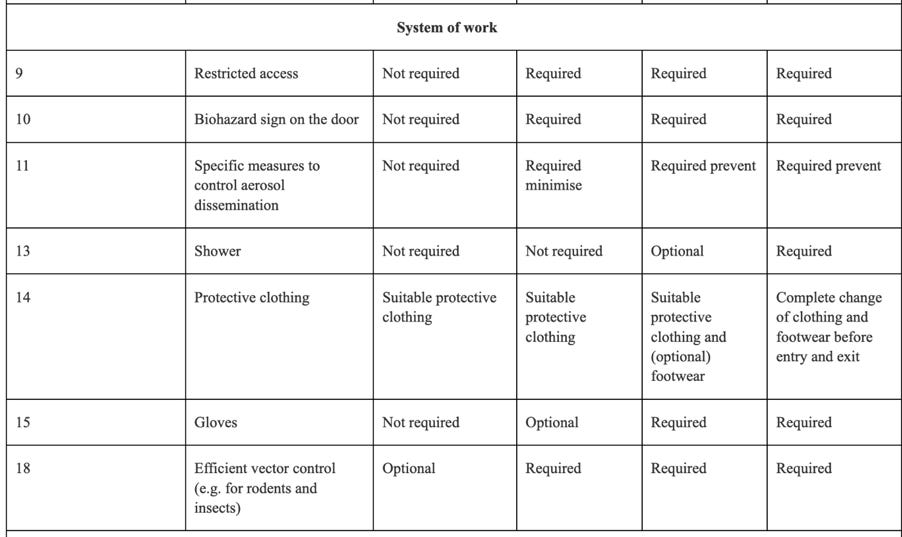
Clearly circumstances could arise where Directive 2009/41/EC needs to be followed in the context of human Clinical Trials, for instance, in respect of a Covid-19 medicine containing GMOs. One would hope any such Clinical Trial involving Directive 2009/41/EC and a large number of participants, required only Containment 2 Level measures or lower, but of course that would depend upon the identified risks in respect of the GMOs being handled and administered.
Directive 2009/41/EC differs critically from Directive 2001/18/EC in so far that Information required to be submitted to a competent authority before a sponsor can commence dealing with a GMO in lab setting, is information the general public has no automatic right to receive. Public access to that information is discretionary, and even if access is granted, that does not mean access to all the GMO information submitted by the sponsor, as seen under Article 12:
Where a Member State considers it appropriate, it may provide that the public is to be consulted on aspects of the proposed contained use
Having regard to the research efforts and activities of pharmaceutical companies since at least 2020 and beyond with respect to SARS-CoV-2, including Clinical Trial research and the 10s of 1,000s of participants in Covid-19 Clinical Trials too numerous to list, this discretion whether to allow the general public or indeed Clinical Trial participants access to, or knowledge of, any GMOs being used in those Clinical Trials would be a cause for alarm, yes?
Regulation 2020/1043
Notification Information for GMOs No Longer Required
- The Covid-19 Drugs Exclusion -
In the foregoing pages an attempt was made to abridge and present the voluminous information required from sponsors about the Genetically Modified Organisms they seek to either release into the environment, or place into products for the market, or deal with in a laboratory setting, where the latter can involve Clinical Trials.
Except for laboratory settings under Directive 2009/41/EC, there has existed a high degree of disclosure required to the general public of GMO information needing to be submitted to authorities prior to environmental or product releases.
The right of the public To Be Informed and submit responses and comments to authorising bodies of government, and to carry on public discourse and debate towards any proposed GMO activities or products, must be regarded as an unimpeachable and entrenched Human Right over and above any stated legal right, in so far that GMOs quite simply threaten to alter Human DNA forever, and thereby the Identity or Existence of Human Beings, let alone considerations of the immediate threats to Human Health GMOs pose.
European Parliament Regulation 2020/1043 illegally suspended that Human Right in 447.7 million European citizens in July of 2020, being a Human Right each of those 447.7 million people shared with the other 7.4 billion people of Earth in 2020. The Human Right To Be Informed about any GMO that could enter their bodies.
Let us recall what our forefathers foresaw and sought to protect us from.
First, European regulators and all other medicines regulators aided by complicit politicians turned their backs on Articles 16, 20, and 28 (amongst others) of the Universal Declaration on Bioethics and Human Rights, a ‘universal’ expression of values and protections given lip service when medicines and genetics regulators said they would stand behind the Declaration forever:
Protecting future generations
The impact of life sciences on future generations, including on their genetic constitution, should be given due regard.
Risk assessment and management
Appropriate assessment and adequate management of risk related to medicine, life sciences and associated technologies should be promoted.
Denial of acts contrary to human rights, fundamental freedoms and human dignity
Nothing in this Declaration may be interpreted as implying for any State, group or person any claim to engage in any activity or to perform any act contrary to human rights, fundamental freedoms and human dignity.
Next, and the great International Covenant on Civil and Political Rights (ICCPR) has also been thrown to the wind. Article 7 could not have been more clearly stated nor more relevant:
No one shall be subjected to torture or to cruel, inhuman or degrading treatment or punishment. In particular, no one shall be subjected without his free consent to medical or scientific experimentation.
Once upon a time 173 countries promoted themselves as being a ‘State Party’ to the ICCPR … it was nothing but theatre for most.
A Human Right denied to One is a Human Right denied to All.
This Human Right remains suspended in Europe in respect of any GMO information relating to medicinal products intended to treat Covid-19, where used in a laboratory or Clinical Trial setting.
What GMO Information has Regulation 2020/10423 withheld?
Under Article 2 of Regulation 2020/1043 any Clinical Trial involving an investigational medicine intended to treat or prevent Covid-19, from July of 2020, was no longer required to submit the GMO information previously required under Part B of Directive 2001/18/EC, (Articles 6 to 11), nor submit GMO information under Articles 4 to 16 of Directive 2009/41/EC (GMO containment in a lab setting), with the result being for the sponsor(s) of Clinical Trials:
1. No need to submit an Environmental Risk Assessment for the GMOs proposed to be used and administered to Clinical Trial participants.
2. No need to wait for any approval from any authority to handle and administer GMOs to participants in Clinical Trials.
3. No need for any authority to inform or consult with the public before the proposed handling and administration of GMOs to participants in Clinical Trials.
4. Consequently, no need for sponsors of Clinical Trials to inform participants the investigational medicine to be administered to them, contained GMOs.
5. No need to evaluate the risks posed by the GMOs to be administered in a laboratory setting, which is inclusive of Clinical Trials, therefore no evaluation of the Class of risk posed by the GMO was required, nor an assessment of the Level of Containment required by laboratories dealing with GMOs being investigated for Covid-19, therefore not even Biohazard signs were required to be displayed.
Not only does/did Article 2 of Regulation 2020/1043 deny the public and particularly Clinical Trial participants any knowledge of GMOs being administered, and completely abrogate all notions of participants being fully informed for the purposes of Informed Consent, the complete absence of any requirement to undertake any risk assessment in respect of GMOs proposed to be used, in fact went one step further, and dispensed with the need for any sponsor to even question whether an ingredient even met the legal definition of a Genetically Modified Organism.
As all requirements to notify authorities of any substance that could be deemed a GMO had been suspended, sponsors were freed to unleash all their experimental stocks of GMOs no matter what the previous risk assessment may have been on those stocks, and no matter what Level of Containment they once may have been confined to, so long as a sponsor invoked Regulation 2020/1043. For instance, highly experimental and high risk GMOs previously designated for Contained Use to say a Level 4 Containment facility, could now be brought into any laboratory environment and unbeknownst to Clinical Trial participants, administered by staff with minimal qualifications, with little to no obligations on the sponsors to closely monitor all the known and possible adverse effects from those GMOs.
Under Article 2(2) all ‘foreseeable negative environmental impacts’ resulting from the intentional release by sponsors of investigational products containing GMOs into the environment, became the sole responsibility of those sponsors ‘to minimise’. Recall, ‘environment’ is inclusive of individual human bodies. How possibly could any sponsor or manufacturer ‘minimise’ negative impacts upon Clinical Trial participants, after those participants had been administered genetically modified organisms?
Under Article 2(3), should a sponsor of Covid-19 Clinical Trials involving GMOs subsequently seek Market Authorisation for their investigational drug to go into the public marketplace, any such application since July of 2020 has not been required to furnish any consents from authorities for the use and administration of GMOs in a prior Covid-19 Clinical Trial. Authorities were freed of any responsibility with respect to GMOs used in any Covid-19 Trials.
However, where GMOs were used in Covid-19 Clinical Trials and the sponsor subsequently applied for marketing authorisation of a product from those Trials containing GMOs, Article 2(3) did not exempt any sponsor from providing the GMO information items discussed above in Annexures II, III, and IV, of Directive 2001/18/EC.
Part C of Directive 2001/18/EC would not apply (Articles 13 to 24) to applications for Covid-19 products containing GMOs, so long ‘as they are authorised by Community legislation which provides for a specific environmental risk assessment carried out in accordance with the principles set out in Annex II and on the basis of information specified in Annex III’ of Directive 2001/18/EC.
That other Community legislation requiring the same GMO information as seen under Annexures II, III, and IV of Directive 2001/18/EC is seen in the case of the Pfizer and Moderna Covid-19 applications, under Annex 1 of Directive 2001/83/EC, specifically Part 1.6, where the provision of the Annex 1 information is an obligation of sponsors pursuant to Article 8(3) of Directive 2001/83/EC, which was the application pathway used by each of Pfizer and Moderna.
Critically, the requirement to Inform the public of GMO ingredients was preserved by this application pathway (see Part 1.6, paragraph 4, indent 6).
But – no such GMO information was supplied by either Pfizer or Moderna. In the case of Moderna, the CHMP EPAR setting out the Article 8(3) and Annex 1 information used to authorise the Moderna Covid-19 drug, simply noted at page 37 in respect of GMOs:
Not applicable.
The question becomes, Why?
And that question can only be answered by recourse to the European definitions for what constitutes a Genetically Modified Organism, and whether CHMP and EMA properly considered those definitions for the Pfizer and Moderna products.
Recall first that each of the Pfizer and Moderna applications recognised their products came under Article 3(1) of Regulation (EC) No 726/2004, (see Pfizer at page 8 HERE and Moderna at page 9 HERE), being specifically a type of medicine shown at Part 1 of the Annex of Regulation (EC) No 726/2004:
Medicinal products developed by means of one of the following biotechnological processes:
- recombinant DNA technology,
- controlled expression of genes coding for biologically active proteins in prokaryotes and eukaryotes including transformed mammalian cells,
- hybridoma and monoclonal antibody methods.
For the Pfizer and Moderna the first indent above defines their Covid-19 products: Medicinal products developed by recombinant DNA technology. To be clear, recombinant DNA technology is used to create the modified RNA which is the main ingredient of their products.
More correctly those products contain LNP-modRNA complexes, as the mRNA/modRNA derived from the recombinant DNA technologies is incapable of transfecting human cells without first being encapsulated within lipid nanoparticles, or LNPs.
To be a GMO, or not to be a GMO
It is time now to ask the question: Do the Pfizer and Moderna Covid-19 ‘vaccines’ contain GMOs?
In short: Yes, they do.
The guiding rule when answering the question is to stick to the legal definitions of a GMO, as they are the only definitions that count in a Court of Law. Many an expert in the biological sciences have views, and many have differing views, for what constitutes a genetically modified organism, but those experts must defer to the legal definitions and restrain themselves to only the words contained in those definitions. Elaborations on those words and terms takes one outside of a stated definition and introduces only more considerations not found in the original definition, set down in law.
We return now to Article 2 of Directive 2001/18/EC containing the definition for what constitutes a GMO (emphasis added):
(1) "organism" means any biological entity capable of replication or of transferring genetic material;
(2) "genetically modified organism (GMO)" means an organism, with the exception of human beings, in which the genetic material has been altered in a way that does not occur naturally by mating and/or natural recombination;
Within the terms of this definition:
(a) genetic modification occurs at least through the use of the techniques listed in Annex I A, Part 1;
Annex I A provides examples of techniques for genetic modification and is not exhaustive as indicated by the term ‘inter alia’. Regardless, we already find a technique described at Annex I A, Part 1(2), that is sufficient for proving the point:
TECHNIQUES REFERRED TO IN ARTICLE 2(2)
PART 1
Techniques of genetic modification referred to in Article 2(2)(a) are inter alia:
(2) techniques involving the direct introduction into an organism of heritable material prepared outside the organism including micro-injection, macro-injection and micro-encapsulation;
In a nutshell we need only satisfy the following elements when looking at the active ingredient contained in the Pfizer and Moderna products, namely, any biological entity that can be said:
1. Is capable of transferring genetic material; and
2. The genetic material has been altered in a way that does not occur naturally; and
3. Arose as a consequence of genetic modification techniques which introduced ‘heritable material’ prepared outside the biological entity.
Of course the candidate here is the LNP-modRNA complex found in each of the Pfizer and Moderna products.
At this point it is appropriate to introduce the Australian theme to this discussion.
Australia also has its laws governing the authorisation and management of genetically modified organisms under the Gene Technology Act 2000. When we look to that legislation we see very similar provisions when it comes to defining a GMO. The definitions are found under Section 10 (in part, relevantly):
"organism" means any biological entity that is:
(a) viable; or
(b) capable of reproduction; or
(c) capable of transferring genetic material.
"genetically modified organism" means:
(a) an organism that has been modified by gene technology
..
"gene technology" means any technique for the modification of genes or other genetic material ..
So the Australian criteria to be satisfied when looking at the Pfizer and Moderna products are whether any biological entity:
1. Is capable of transferring genetic material; and
2. The biological entity was modified by gene technology; and
3. The gene technology involved any techniques for the modification of genes or genetic material.
Preliminary question: Can the LNP-modRNA complexes properly be called ‘any biological entity’?
The short answer is Yes. This is not a controversial answer, as what can be called a ‘biological entity’ encapsulates so much when left unrestricted by the preceding ‘any’.
Next and key to both the Australian and European definitions: Is the LNP-modRNA capable of transferring genetic material?
The short answer is Yes. For the long answer I provide the following explanation supplied to me from a PhD in Molecular and Cellular Biology who was asked the very same question. This good Doctor also consulted with several similarly qualified colleagues, one with a PhD in Genomics, before responding:
The LNP acts as a transfectant, which enables the delivery of the modRNA across the
membrane of human cells into the cytoplasm of cells. The design of the LNP also assists in the release of the modRNA from the endocytic compartment and into the cytoplasm. In addition, Sattar et al. have detected both spike mRNA and spike protein in the nucleus of cells (Sattar et al 2022). While the exact mechanism of this process is not fully defined, it has been suggested that the spike protein contains a “nuclear localisation signal” in its sequence, which allows for its transport into the nucleus. The authors propose that the spike mRNA can bind the spike protein, thereby “hitchhiking” a ride to the nucleus. The extent of function of the RNA in the nucleus is unknown. This calls into question, the very vehement claims that the Pfizer and Moderna modRNA products do not enter the nucleus.
It is important to note that a previous study demonstrated that the Pfizer modRNA (BNT162b2) can be reverse-transcribed in an in vitro cellular system (Alden et al 2022). This process converted the modRNA to DNA, which is the form of genetic material that is passed on from generation to generation. Together, these studies place the LNP-modRNA complex as an agent for the transfer of genetic material.
Next is the question: Were (are) the LNP-modRNAs modified or created by gene technology?
The European definition asks the question from a slightly different angle namely, were the LNP-modRNAs altered in a way that does not occur naturally. These questions were also asked of the same PhDs who first answered Yes, gene technology is involved, then further explained:
Firstly, the sequence that encodes the entire modRNA is a fusion of several different sequences, which include a 5’-cap, a 5’-UTR, derived from the human alpha-globin gene, along with an optimised Kozak sequence (to assist in driving robust expression), a codon-optimised coding sequence (different from the original SARS-CoV-2 sequence), a 3’-UTR at the end, consisting of two other human sequences, and a poly-adenosine tail (to assist with stability of the mRNA) (Nance’21).
Once the modified DNA was created to include all the above mentioned features, the manufacturers utilise in vitro transcription (IVT) to create the modRNA molecules (Nance et al 2021). During this process, they provided the M1Y in the mix, rather than the original Uracil. This rendered the resulting modRNA molecules as highly modified versions compared with the SARS-CoV-2 mRNA encoding the spike protein.
The above response from learned doctors in the fields of molecular and cellular biology and genomics, clearly satisfy deeming the LNP-modRNAs contained in the Pfizer and Moderna products as Genetically Modified Organisms.
The European definition does ask one further question being whether the genetic modifications introduced ‘heritable material’ prepared outside the LNP-modRNA.
From the lab process described above we can acknowledge the constituent elements and final construct of the modRNA are a creation of sophisticated gene technology processes, involving manufacturing steps that first produce the synthetic modRNA, then subsequent steps that result in large vats of that modRNA bound within LNPs.
But in answer to the European issue of ‘heritable material’, we have already seen reference above to the peer reviewed paper by Alden et al 2022 evidencing Pfizer’s modRNA being reverse transcribed into human DNA. That process evidences the modRNA to be composed of ‘heritable material’, being material able to alter human DNA. This finding by Alden et al was further confirmed by the findings of Qin et al 2022 evidencing mice who received LNP-modRNA acquired certain immune traits, but most importantly were able to evidence:
“.. mice pre-exposed to the mRNA-LNP platform can pass down the acquired immune traits to their offspring ..”
The above statement is conclusive of the LNP-modRNA complex changing DNA, being changes passed along to their offspring – that can only involve earlier ‘heritable material’ received by the parents.
This is everything we were told to fear from GMOs, from Genetically Modified Organisms, yet both the European and Australian regulators failed to discuss the possibility of these biological entities, these modRNAs, designed and constructed using the very elements that constitute DNA, as possibly being GMOs, despite past knowledge (let alone the precautionary principle) indicating before anything else, they should have been deemed Genetically Modified Organisms until proven otherwise.
Let us be clear on the point. The paper by Alden et al showing Pfizer’s product reverse-transcribing was not just a chance set of findings. Markus Alden together with the rest of the team - Francisko Olofsson Falla, Daowei Yang, Mohammad Barghouth, Cheng Luan, Magnus Rasmussen, and Yang De Marinis – that team all knew about mRNA and its ability to reverse-transcribe into human DNA. And that prior knowledge has been available to all global medicines regulators for quite some time, well in advance of the Pfizer and Moderna Covid-19 applications.
That knowledge and information has been the ‘daily bread’ of the regulatory experts charged to detect and question whether a new biological entity just like the modRNAs should properly be seen as likely GMOs, as soon as Pfizer and Moderna walked in their doors to discuss lodging applications for market authorisation. For reference, that prior knowledge on mRNAs is expertly catalogued in the peer-reviewed paper by Associate Professor Domazet-Loso titled mRNA Vaccines: Why Is the Biology of Retroposition Ignored? (April 2022) which will be drawn upon in detail below. For those who find the science jargon a difficult read I invite you to watch the video by Domazet-Loso where he steps the viewer through his paper in easy terms, HERE.
So let us deal with the one essential claim by Pfizer and Moderna: modRNA does not enter the cell nucleus and does not interact or integrate with the genome.
Both companies have made this claim the world over. It must be immediately noted that neither Pfizer nor Moderna provided any scientific basis for this claim. That should have been the sole red flag for regulators everywhere to ask: How do you know Pfizer, Moderna?
Retroposition
The true state of affairs – mRNAs do Integrate.
Associate Professor Domazet-Loso is the only guide we need:
I was not able to track down any experimental or theoretical study that specifically addresses the possibility of genome integration of mRNA therapeutics.
This shortage of relevant studies is reflected in numerous reviews [4,5,6,9,10,14,15,16,17,18], book chapters on the mRNA vaccines [13,19,20,21,22] and documents of international organizations [23,24,25], which often state that mRNA vaccines do not pose the risk for genome integration but do not cite any references in support of this idea.
Many of them simply state that vaccine mRNA cannot integrate into the host genome without explaining why this is not possible [3,10,12,19,20,21,22,26,30].
Domazet-Loso notes that none of these reviews, chapters, or publications by international organisations, (typically the WHO), ever bothered to reference the established knowledge on the biology of Retroposition, seen for instance in peer-reviewed papers in 2009, 2017, 2017, 2020, 2021. These 5 peer-reviewed papers each cite anywhere from 44 to over 130 other earlier peer-reviewed papers on the subject of Retroposition, reaching back into last century. Those papers in turn cite many more Retroposition papers, with many leading back to the 1950 discovery of genetic transposition by Barbara McClintock, which won her the Nobel Prize in 1983. Thus it is beyond question - the field of Retroposition studies was well established before Pfizer and Moderna stepped-up with their products. Domazet-Loso continues:
In many eukaryotes [organisms whose cells have a nucleus], the cellular mRNAs of various genes are endogenously reverse-transcribed and reintegrated into the genome, yielding their retrocopies ..
.. the estimated number of retrocopies in the human genome varies, but the figures in most studies are approximately 8000; these retrocopies are derived from around 2500 parental genes, i.e., genes whose mRNAs are reverse-transcribed and integrated into the genome ..
.. mRNA retroposition also occurs in somatic tissues [other tissue about the body] .. it is known to be common in cancer tissues [62,77,78,79,80] and to occur during early development [68,69]
.. The mechanism that leads to the formation of retrocopies in a human lineage is relatively well studied and predominantly includes Long Interspersed Element-1 (L1/LINE-1) retrotransposons [36,38,40,46,81]
At this point some will seek to seize upon the WHO statement: “The only known mechanism by which RNA can integrate into the host genome is in the presence of a retrovirus particle containing reverse transcriptase.”
Domazet-Loso was aware of this statement too, stating: ‘the vaccinology field, for an unclear reason .. [is] .. unaware of the existence and significance of L1-driven retroposition in humans.’ This veiled sarcasm is appropriate and a shot-over-the-bow indictment. Yes, every reader should question why the WHO continues to remain so critically unaware of scientific literature dating back decades.
The rebuttal to the WHO statement is simple – integration into the host genome requires reverse transcriptase. The Long Interspersed Element-1 (L1) mRNA abundantly within humans codes for ORF1 and ORF2 proteins. ORF2 is a reverse transcriptase.
So let us not fool around anymore – the WHO well knows the science of Retroposition in humans but has refused to address the subject as that would involve an implicit admission, albeit an outright logical step, that we are dealing with GMOs when it comes to the Pfizer and Moderna drugs. And the same goes for every medicines regulator about the globe. Every one of them said nothing about Retroposition at the time of the applications, so everyone one of them is doing the triple-down of the double-down denial narrative – in the face of their decades of scientific knowledge of the biology of Retroposition.
Returning to Long Interspersed Element-1 (L1/LINE-1) retrotransposons, and Domazet-Loso continues with the abundant science the WHO and regulators ignore, and Pfizer and Moderna would have you believe they did not know about:
The mere presence of numerous vertically inherited L1 elements, non-autonomous mobile elements and retrocopies in human genomes provides direct evidence that their mobilization repeatedly occurs in the germline [99]
.. The current data suggest that L1 elements show expression and retroposition activity in testes [96,105,106,109], spermatozoa [110,111], ovaries [105,106], oocytes [112] and early embryos [97,99,105,107,108,113]
.. L1 elements .. should be considered an endogenous mutagen in somatic tissues [99,100,106,114]. L1 elements are expressed in diverse human somatic tissues, including liver, spleen, adrenal glands, lungs, heart and brain [106]; lymphoblastoid cell lines [115]; platelets; megakaryocyte; and T cells [98]. Expression and retroposition activity of L1 elements was detected in vascular endothelial cells as well [109,116]. However, somatic L1 retroposition has been extensively studied only in the brain, cancer tissues and the gastrointestinal tract [45,78]
.. L1 retroposition occurs in diverse cell types of the central nervous system, including glial cells, neuronal progenitor cells, differentiating neurons and mature non-dividing neurons [118,121,123,124,125,126]
In respect of the modRNAs contained in the ‘vaccines’, Domazet-Loso then set about addressing their capability for being reverse-transcribed, opening with:
Evidently, various mRNAs in humans could be reverse-transcribed and integrated into the genome via L1 retroelements with negative effects on fitness. However, this does not readily imply that this will occur to vaccine mRNAs. A definitive answer will come from experiments and population monitoring ..
Two issue arise with the above.
Firstly, Domazet-Loso would not have mentioned the need for ‘experiments’ had he seen the earlier paper by Alden et al 2022 before seeking publication (he subsequently did, but too late to alter his paper). The Alden paper would have saved Domazet-Loso going into great detail to show the significant probability of reverse-transcription with the modRNA drugs. Alden et al proved all the points Domazet-Loso put forward. In a nutshell as Domazet-Loso states at 27:48 mins in the video presentation of his findings:
“.. all applied [genetic] engineering solutions [involved with the modRNAs] point that the vaccine mRNA molecules are, intentionally or unintentionally, designed to be integrated into genomes as easily as possible”.
Secondly, and perhaps more importantly, under the legal definitions applicable in both Europe and Australia, reverse-transcription was not, in and of itself, a necessary precondition for categorising these modRNAs as GMOs.
The fact the LNP-modRNA complex enables entry into a cell (just into the cytoplasm) is sufficient. Due to the fact of this entry alone all the legal obligations for performing a detailed GMO Environmental Risk Assessment arose, requiring for instance, an assessment of the possibility of subsequent entry into the nucleus and the possibility of reverse-transcription, let alone Genotoxicity and Carcinogenicity studies. Those discrete safety assessments were readily able to be performed by Pfizer and Moderna, and for regulators to ensure they performed them, just like Adlen and his team did independently, nearly two years after the release of these products into the bodies of people globally.
Once the Alden et al paper was published in February 2022, regulators globally should have immediately suspended the modRNA products, finally stopping their collusive silence on the issue of these drugs being GMOs, and instead started looking at solutions for how to neutralise the modRNA received by billions of people.
The depth of the infiltration of the Pfizer drug per shot was calculated by Domazet-Loso .. when reading the below double, triple, or quadruple the numbers for second and subsequent doses:
If we ignore the loss of vaccine mRNAs on the route to the cytosol [cytoplasm, just after cell entry] and assume their homogenous distribution among roughly 3 × 1012 nucleated cells in the human body [153], then every nucleated cell could receive about 26 mRNA copies. This is a substantial amount if compared to the expressed human protein-coding genes that have on average 25 mRNA copies per cell [154]. These values show that the quantity of vaccine mRNA delivered in a single dose of BNT162b2 is large enough to theoretically reprogram the transcriptome [the full range of mRNA expressed by cells] of every single human cell that in principle can undergo retroposition.
Regulators and Pfizer and Moderna knew and know these numbers. When Alden et al published their paper, the regulators, Pfizer, and Moderna knew the first steps of an iatrogenic disaster had already been taken. Yet everyone continues to remain silent while Excess Deaths explode in every country these drugs have been released in, concurrently with ever rising ‘vaccine’ injuries, turbo cancers, significant new autoimmune diseases, miscarriages, reproductive issues, and falling birth rates. And do not be mistaken – these adverse health outcomes are just beginning.
Instead second shots, a booster, and second boosters have been pushed by governments and regulators at pace, with now our youngest citizens from 6 months old and pregnant mothers being added to the list of victims and injured, within whom Retroposition mediated genome integration is most active. The repeated pushing of shots only increases the chances of integration into the human DNA library, altering what it means to be indeed, human. Domazet-Loso makes especial note of this fact and treats it in detail, showing how the robust engineering of these modRNAs with their ability to evade normal processes that destroy natural RNA, naturally, essentially provide modRNAs further time and opportunity to ensure genome integration:
.. it is clear that the sequence and codon optimization of vaccine mRNAs increases their functional half-life with an aim to improve their translation efficiency [6,10,27,56,138,139]. Undoubtedly, this prolonged functional half-life increases the chances that vaccine mRNAs encounter L1 machinery and eventually retroposed into the genome.
And all this engineering placed into the bodies of humans come with consequences humanity should not have been forced to face. As Domazet-Loso observes, we face a new frontier and much reckoning:
Our cells evolved under mutational pressure that came from the activity of L1 elements that generate retrocopies of our native genes [37,40]. However, the transfection of human cells with exogenous and artificially modified mRNAs, which have the potential to be retrocopied into the genome (Figure 1C), extends the standard mutational sequence space to the realm of transgenic modifications. It is rather clear that any possibility of transgenesis in humans has ethical concerns that should be properly addressed. This raises two questions: Who is responsible for testing the likelihood of vaccine mRNA retroposition, and who will be responsible for eventual genome modifications resulting from the application of emergency-use mRNA vaccines? The answers to these questions are, without doubt, of outstanding importance for society at large.
The takeaway item is of course ‘transgenic modifications’ in humans as a consequence of modRNAs, where a quick google of transgenic returns:
.. an organism or cell whose genome has been altered by the introduction of one or more foreign DNA sequences from another species by artificial means. Transgenic organisms are generated in the laboratory for research purposes.
In the above we simply replace ‘foreign DNA’ with ‘modRNA retrocopies’ via Retroposition.
We now stand upon a transgenic frontier the making of regulators everywhere, and the WHO. Make no mistake, every modern country has its gene technology experts charged with keeping medicines regulators up-to-speed with all things genetically modified, or that should be deemed to be genetically modified organisms, until proven otherwise.
In Australia that failed government agency is the Office of the Gene Technology Regulator, staffed as it is with Australia’s acknowledged experts, was meant to step-in and hold Pfizer and Moderna to account, by first having them submit and assist complete a Risk Assessment and Risk Management Plan, just like AstraZeneca was required to do with its Covid-19 drug (despite that too being another failure by the OGTR).
The European Medicines Agency failed humanity too when it turned a blind eye to the Pfizer and Moderna Covid-19 GMOs. Clearly a corruption continues to transpire. The depths of coordination and complicity were and remain extraordinary but was achievable with sophisticated global communications and long compromised political structures that long ago stopped serving the people for anything, but fear.
The closing gentle observations by Associate Professor Domazet-Loso raise nothing but appropriate cynical implications:
The mRNA vaccinology field started its development more than 30 years ago [11,31] and L1 retroelements in humans have been studied for more than 40 years [205,206] but obviously without any crosstalk between the two fields ..
.. Unfortunately, all of this creates an impression that L1-driven retroposition is a kind of taboo topic in mRNA vaccinology.
When new drugs constructed with the elements of our genes are introduced seemingly out of nowhere, and within months are scaled-up for a global population told ‘everything is fine, just take it, or else’ .. the failure to acknowledge over four decades of science speaking to the genetic risks associated with the drugs was never an oversight .. it involved concerted planning and intention, involving a criminal avoidance of the real science at all costs.
The implications are now staggering ..
.. the Canaries in our DNA mine are screaming.
End.
Onwards!
Please subscribe, or donate a coffee (I drink a lot of coffee) - “God Bless You!” if you can’t or don’t want to contribute. Coffee donations here: https://ko-fi.com/peterhalligan
THIS: Evidently, various mRNAs in humans could be reverse-transcribed and integrated into the genome via L1 retroelements with negative effects on fitness. However, this does not readily imply that this will occur to vaccine mRNAs. A definitive answer will come from experiments and population monitoring .
THIS IS UNFORGIVABLE..
Thanks PH. Fearsome amount to wade through. That all of the grammar could be addressed in just one word. Probably too blunt? GENOCIDE. Pre meditated. Pre planned. Now in full force. How in the face of such scientifically proven fact that the injections DO cause irreversible, life changing HARMS. Then the actual DEATHS caused. Any government on earth can still inject their own citizens?